A vasculite por imunoglobulina A (IgAV), anteriormente chamada de púrpura de Henoch-Schönlein, é uma vasculite sistêmica por imunocomplexo que afeta predominantemente pequenos vasos.
É a vasculite mais comum da infância, com uma incidência anual de 3 a 26 casos por 100.000 crianças.
Em adultos, a doença é mais rara, com uma incidência anual de 0,1 a 1,8 por 100.000 indivíduos, sendo mais comum em homens, com uma proporção de 1,5 caso homem / mulher (em crianças não há diferença entre os sexos), com maior incidência na quinta e sexta décadas de vida.
A deposição de imunocomplexos de IgA nos órgãos-alvo desempenha um papel central na patogênese da doença.
A doença no adulto tem um risco maior de envolvimento sistêmico, de ser mais grave e recidivante, quando comparado com a doença na criança. O envolvimento renal e o envolvimento gastrointestinal são as principais causas de morbidade e mortalidade em adultos.
Os estudos são escassos e não há critérios classificatórios bem definidos para IgAV, além disso as recomendações de tratamento também não são padronizados.
Patogênese e etiologia
Fatores ambientais e genéticos são resposáveis por anormalidades da imunidade inata e adquirida e no papel primário dos imunocomplexos contendo IgA1 deficiente em galactose em sua patogênese.
A característica patológica mais marcante da IgAV são os depósitos de imunocomplexos contendo IgA1 deficiente em galactose (Gd-IgA1) na parede do vaso. Há uma alteração na glicosilação da região de dobradiça da IgA1, produzindo uma IgA1 aberrante e deficiente em galactose. Três enzimas são críticas na glicosilação de IgA1 e variações em sua atividade ou expressão podem levar à deficiência de galactose em IgA. As citocinas (IL-6 e IL-8) regulam essas enzimas e polimorfismos genéticos dessas enzimas que estão envolvidas na síntese de Gd-IgA1.
Os níveis séricos de IgA estão elevados devido ao aumento da produção e defeitos em sua depuração.
A imunidade da mucosa, especialmente dos órgãos linfoides gastrointestinais, teria um papel fundamental na patogênese.
Foram descobertos loci de suscetibilidade genética para IgAV: antígeno leucocitário humano (HLA)-35, alelos HLA-DRB1*01 e particularmente HLA-DR1*0103. Polimorfismo foi encontrado em genes que codificam citocinas (IL-1, IL-6, IL-8) e em genes que codificam enzimas, afetando a síntese IgA1. Existe uma associação entre a mutação do gene da Febre Familiar do Mediterrâneo e IgAV na população do Mediterrâneo.
Os neutrófilos predominam na infiltração inflamatória em biópsias de pele e gastrointestinais em IgAV. Nos neutrófilos, a reticulação induz a liberação de armadilhas extracelulares de neutrófilos (NETs) e leucotrieno B4 (quimioatraente de neutrófilos), aumentando o dano.
A elevação dos fragmentos C3a, C5a e os depósitos de C3 e C5-9 indicam ativação da via alternativa. Há ativação de linfócitos T CD8 citotóxicos que contribuem para lesão renal em nível glomerular. O TNF-α sérico está elevado e está associado à gravidade da doença.
Infecções e medicamentos são eventos precipitantes bem descritos.
Os patógenos relacionados a IgAV são: Streptococcus do grupo A (o mais frequente), o vírus da parainfluenza, o parvovírus humano B19, o citomegalovírus , o HBV, o HCV, a Helicobacter pylori, a COVID-19.
Os antibióticos, particularmente os beta-lactâmicos, foram considerados os medicamentos mais comumente implicados na predisposição.
Vacinas, bloqueadores do fator de necrose tumoral α, anti-hipertensivos, anticoagulantes, anti-inflamatórios não esteroides e outros analgésicos também foram associados. A glomerulonefrite é menos comum em casos induzidos por medicamentos
Pneumonia, pielonefrite, epididimite e celulite foram encontradas em adultos 4 semanas antes do diagnóstico de vasculite por IgA.
A relação entre vasculite por IgA em adultos e malignidade subjacente é difícil de aferir, com uma relação de 2,5%-10%. A maioria dos cânceres associados são de órgãos sólidos, principalmente mucosa. Níveis sérios mais altos de IgA merecem uma atenção especial.
Clínica
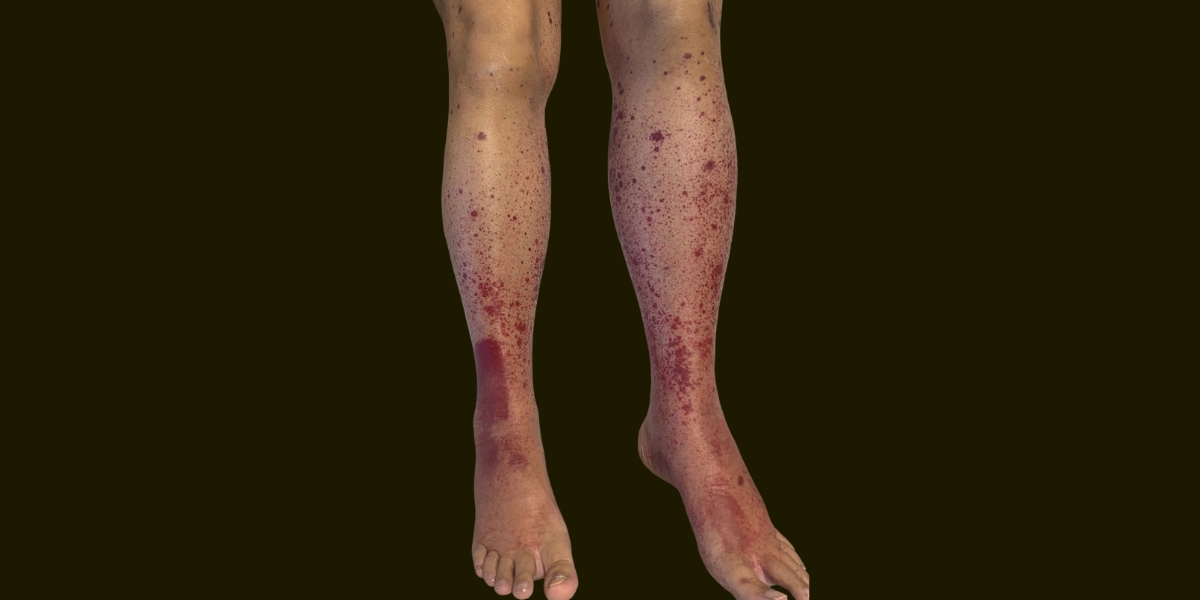
Inclui principalmente manifestação cutânea ( púrpura palpável ), artralgia, dor abdominal (enterite) e glomerulonefrite.
Podem se desenvolver ao longo de dias a semanas e podem variar em sua ordem de apresentação.
A manifestação cutânea ocorre em 96–100% dos casos na apresentação, envolvimento articular em 61% e intestinal em 48-53% dos casos ao diagnóstico. Até 30% dos pacientes podem ter alteração de função renal.
As lesões cutâneas geralmente se apresentam como lesões eritematosas, maculares evoluindo para petéquias/púrpuras. Sendo mais comum nos membros inferiores, porém pode se extender para todo o corpo, podendo evoluir para bolhas, pústulas e púrpura necrótica ou hemorrágica. As lesões podem ser assintomáticas, dolorosas ou pruriginosas. Na biópsia é encontrado vasculite leucocitoclástica típica afetando principalmente os pequenos vasos superficiais, com depósitos predominantes de IgA. A depender do tempo da biópsia o depósito por ser negativo, porque a IgA pode se degradar em lesões necróticas mais antigas. Os critérios EULAR sugerem a avaliação com coloração por hematoxilina e eosina nas lesões estabelecidas com uma biópsia lesional por punch (24h a 1 semana) não necróticas; e para avaliação de imunoflorescência biópsia lesional ou perilesional por punch em lesões não necróticas iniciais (24-48h).
O envolvimento articular geralmente é transitório, não deformante, sendo mais comum nas articulações dos joelhos e tornozelos. Foi relatado em cerca de 30%-60% dos casos.
O sintomas gastrointestinais (GI) foram relatados em cerca de 37%-65% dos casos em adultos. Se apresentam geralmente como dor abdominal, naúseas, vômitos, podendo evoluir com sangramento intestinal. Esses sintomas se devem ao edema intestinal, isquemia intestinal e hemorragia de submucosa. Podem ocorrer complicações como intussuscepção, infarto e perfuração. Pancreatite e acometimento da vesicula biliar são manifestações mais raras. A mortalidade por esse acometimento é raro. Qualquer evidência de envolvimento GI seja investigada com tomografias computadorizadas e/ou endoscopias. Se não houver envolvimento GI, os pacientes devem monitorar esses sintomas e relatá-los imediatamente se identificados.
A manifestação renal mais comum é a hematúria microscópica associada a proteinúria, cerca de 33% podem apresentar alteração de função renal. Hipertensão é tipicamente observada em um terço dos casos. Exames de urina, quantificação de proteína urinária e creatinina sérica devem fazer parte da avaliação inicial, se normais devem ser repetidos a cada 2 semanas no primeiro mês e depois a cada 2 meses no primeiro ano, se alterados o paciente deve ser referenciado a um nefrologista. Alterações que indicam acometimento renal: proteinúria >0,3 g/24 h; hematúria (>5 hemácias/campo de grande aumento) ou cilindros de hemácias .Os fatores de mau prognóstico são uma função renal prejudicada no início, proteinúria > 1 g/dia no início da doença, hematúria macroscópica, hipertensão, proteinúria persistente >1 g/dia durante o acompanhamento, proporção de fibrose intersticial (> 10%), necrose fibrinoide (> 10%) e esclerose glomerular (> 20%). Os achados na biópsia renal em IgAV são semelhantes aos observados na nefropatia por IgA, com uma glomerulonefrite proliferativa com depósito predominante de IgA. O KDIGO sugere a biópsia renal se proteinúria maior ou igual a 0,5g/24h persistente (mais de 4 semanas) ou alteração de função renal sem outra causa aparente. Os tipos histopatológicos de glomerulonefrite (GN) que podem ser encontrados na V-IgA foram classificados por Pillebout et al (classe I GN mesangial; classe II GN segmentar focal; classe III GN proliferativa endocapilar; classe IV GN proliferativa endocapilar e extracapilar; classe V esclerosante). As mais comuns são classe II e III
Outras manifestações descritas: dor testicular, hemorragia alveolar, pneumopatia intersticial e neuropatia periférica.
Na criança geralmente tem um curso mais benigno do que no adulto.
Adultos tem menos manifestações gastrointestinal, porém com mais manifestações renais. Pacientes adultos apresentam recidivas em aproximadamente 20% dos casos.
Diagnóstico
O diagnóstico se baseiam em achados clínicos e anatomopatológicos.
Níveis elevados de IgA sérico são encontrados em 31-60% dos casos, e o seu valor normal não excluiu IgAV. Espera-se FAN não reagente (pode ser positivo em até 14% dos casos), ANCA negativo (até 4% dos casos tem relato de positividade); fator Reumatoide e crioglobulinas negativos; complemento normal. Velocidade de hemossedimentação (VHS) geralmente aumentado.
Os critérios de classificação EULAR/PRINTO/PRES foram validados em um estudo de vasculite por IgA em adultos com uma sensibilidade geral de 99%.
Sintomas constitucionais, como febre, perda de peso e fadiga, não estão incluídos nos critérios EULAR, porém são frequente em adultos.
A púrpura é um critério obrigatórios, e deve ser não relacionada à trombocitopenia. Os demais itens avaliados são: dor abdominal, histopatologia típica (cutânea ou renal), artralgia ou artrite (início agudo) e acometimento renal.
Para atingir os critérios precisa ter a púrpura e + 1 dos quatro itens avaliados acima.
Diagnóstico diferencial
Vasculites associadas a anticorpos anticitoplasma de neutrófilos (ANCA)
Outras vasculites mediadas por imunocomplexos (crioglobulinemia e vasculite urticariforme)
Vasculites limitadas à pele e secundárias a doenças autoimunes sistêmicas, como lúpus eritematoso sistêmico, doença de Sjögren, artrite reumatoide, doença mista do tecido conjuntivo.
Outros: septicemia, coagulação intravascular disseminada.
Recomenda-se que todos os pacientes adultos com IgAV sejam avaliados quanto a causas secundárias ( malignidade, cirrose hepática, doença inflamatória intestinal, hepatite virais e HIV).
Tratamento
O tratamento da doença grave é complexo e controverso, sem evidências claras de que os corticosteroides e/ou imunossupressores melhorem o resultado a longo prazo. Não há ensaios clínicos randomizados que os apoiem, sendo uma extrapolação de tratamentos usados em outras doenças autoimunes sistêmicas ou estudos pediátricos.
A terapia é frequentemente direcionada ao fenótipo clínico, considerando que o curso da doença é frequentemente benigno e a remissão também pode ser espontânea.
Artralgias e manifestações cutâneas leves podem ser tentado inicialmente com sintomáticos.
Em crianças o uso profilático de glicocorticoides sistêmicos em IgAV extrarrenal não reduz a incidência de envolvimento renal, por isso o KDIGO não orienta um tratamento profilático, devido toxicidade potencial deste medicamento e à falta de qualquer benefício claro.
Inibidores da ECA devem sempre ser usados em caso de proteinúria leve a moderada e hipertensão.
Em casos com manifestações de risco de vida ou de órgãos, glicocorticoides sozinhos ou combinados com medicamentos imunossupressores são rotineiramente usados (a maioria dos estudos são em população pediátrica).
Colchicina e dapsona são opção para manifestações cutâneas.
A dapsona com uma resposta boa cutânea, porém com recidiva rápida após a suspensão da medicação. Antes de usá-la, os níveis de glicose-6-fosfato desidrogenase devem ser determinados (sua deficiência pode causar hemólise e metemoglobinemia).
Não há dados de ensaios randomizados comparando terapia de suporte e corticoide na população adulta com IgAV, portanto, o tratamento desses pacientes — particularmente daqueles com envolvimento renal — é parcialmente baseado em ensaios realizados em pacientes com nefropatia por IgA, nesses casos foram o glicorticoide foi protetores contra a deterioração da função renal sem efeitos colaterais notáveis. Na nefropatia por IgA, o KDIGO sugere início de tratamento nos pacientes com proteinúria ≥0,5g em 24h devido risco de declínico de função renal com prednisona 0,5mg/kg/d no max 40mg (kdigo sugere associação com bactrim para profilaxia) com redução após 2 meses de uso de 5mg por mês durante 6-9 meses. Perda de mais 50% da TFG em 3 meses, sinais de crescente na biópsia, recomendação de seguir a recomendação de tratamento de vasculite ANCA.
Para azatioprina não há dados disponíveis sobre o uso em adultos com vasculite por IgA. Em pacientes pediátricos, a azatioprina tem sido usada em associação com corticoide ou outros agentes em casos de nefrite. Os resultados são encorajadores, pois essa combinação parece melhorar o curso clínico da nefrite. Pode ser uma opção também para erupção cutânea persistente, artrite ou envolvimento abdominal.
O metotrexato é outra opção terapêutica que tem sido usada com sucesso como poupador de corticoide, porém também sem dados em adulto. Para criança é uma opção para os casos sem envolvimento renal.
O micofenolato tem sido usado para manifestação renal, com dados também extrapolados por apresentar alterações patológicas renais semelhantes da vasculite por IgA com a nefropatia por IgA. Em pacientes adultos, alguns estudos (retrospectivos) sugerem que o micofenolato pode ser útil no tratamento do acometimento renal na vasculite por IgA (taxa maior e mais rápida de remissão).
A ciclosporina (CsA) também pode ser uma opção para os casos de acometimento renal. Em um relatório de Kalliakmani et al. pacientes com proteinúria nefrótica tratados com CsA associado ao corticoide obtiveram remissão completa ou parcial da síndrome nefrótica e a função renal permaneceu estável durante o acompanhamento de 5 anos. No estudo de Jauhola et al. que compararam CsA com metilprednisolona na IgAV com proteinúria nefrótica ou nefrite crescente, todos aqueles que receberam CsA obtiveram remissão da proteinúria em 3 meses.
Sobre o uso da Ciclofosfamida em adultos, o único estudo prospectivo e randomizado multicêntrico foi conduzido por Pillebout et al. O estudo comparou prednisona em altas doses isoladamente versus prednisona em altas doses mais CYC (seis pulsos de 0,6 g/mq) em 54 pacientes com IgAV comprovado por biópsia e manifestações graves, incluindo glomerulonefrite proliferativa e/ou complicações gastrointestinais graves . Aos seis meses, nenhuma diferença foi encontrada entre os dois grupos na taxa de remissão e, aos 12 meses, o resultado renal, mortes e eventos adversos também não diferiram. Isso sugeriu que CYC não poderia melhorar o prognóstico de IgAV de início na idade adulta. Os dados sugerem que a eficácia do ciclofosfamida na vasculite por IgA de início adulto é incerta, sendo usada ainda nos casos de GN crescente (> 50% dos glomérulos), síndrome nefrótica e/ou insuficiência renal progressiva.
Em relação ao rituximabe, um estudo com 22 pacientes adultos com vasculite por IgA recidivante ou refratária, observou uma taxa de remissão de 91% em seis meses de terapia, um bom resultado renal (apenas um paciente desenvolveu DRC) e uma diminuição acentuada na proteinúria durante o acompanhamento (∼5 vezes em relação ao valor basal). Além disso, o RTX pareceu eficaz em caso de retratamento devido à recidiva e, mais interessante, também quando foi usado como monoterapia. Uma revisão sistêmática também concluiu que o RTX parece ser um agente seguro e útil na indução da remissão da doença e na redução de tratamentos imunossupressores em pacientes pediátricos e adultos com IgAV resistentes ou refratários ao corticoide ou outros medicamentos imunossupressores.
Imunoglobulina sua eficácia foi relatada em dores abdominais ou na redução da proteinúria. Em um estudo prospectivo e aberto com 14 adultos com GN estágio III e função renal normal, Rostoker et al. avaliaram altas doses de IGIV (2 g/kg mensalmente) por 3 meses, seguidas de Ig IM (0,35 ml/kg a cada 15 dias) por 6 meses. Proteinúria, hematúria, índice de atividade histológica e depósitos imunes melhoraram na maioria dos casos.
Na plasmaférese, Augusto et al. relataram 11 adultos com IgAV grave tratados com corticoide e troca de plasma, com melhora rápida e bons resultados a longo prazo. Séries de casos não controlados descrevem seu benefício potencial associado à corticoide para acelerar a recuperação em pacientes com complicações extrarrenais graves.
Outros tratamentos: mudança do estilo de vida (diminuir consumo de sódio se acometimento renal, parar de fumar, controle adequado da pressão em níveis menores ou iguais a 120x70mmhg se acometimento renal); uso de antiproteinúricos se acometimento renal, como IECA/BRA e/ou iSGLT2.
Prognóstico
Na maioria dos casos, a doença é autolimitada e o prognóstico é favorável. O prognóstico a curto prazo depende da gravidade da condição gastrointestinal, enquanto o prognóstico a longo prazo depende da gravidade da nefrite. Os adultos geralmente requerem tratamentos mais agressivos e têm uma taxa de recuperação menor do que as crianças.
Na série de Pillebout et al as causas mais comuns de morte foram neoplasias 27% (pulmão 14%, trato respiratório superior e gastrointestinal 8%), infecções 16%, devido a envolvimento TGI grave 11% e doenças cardiovasculares 9%. Em relação à sobrevida renal, 11% dos pacientes desenvolveram DRC.
Referências
- Federica Maritati, Alice Canzian, Paride Fenaroli, Augusto Vaglio, Adult-onset IgA vasculitis (Henoch-Schönlein): Update on therapy, La Presse Médicale, Volume 49, Issue 3, 2020, 104035, ISSN 0755-4982.
- Kelly, Brenna G. et al. Navigating the initial diagnosis and management of adult IgA vasculitis: A review. JAAD International, Volume 8, 71 – 78
- Ozen, Seza et al. EULAR/PRINTO/PRES criteria for Henoch–Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Annals of the Rheumatic Diseases, Volume 69, Issue 5, 798 – 806
- Pillebout E, Sunderkötter C. IgA vasculitis. Imunopathol Semin. 2021 Oct;43(5):729-738. doi: 10.1007/s00281-021-00874-9.
- Song Y, Huang X, Yu G, Qiao J, Cheng J, Wu J, Chen J. Pathogenesis of IgA vasculitis: an updated review. Frontal Immunol. 2021 Nov 9;12:771619. doi: 10.3389/fimmu.2021.771619.
- Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: outcomes and prognostic factors. J Am Soc Nephrol. 2002 May;13(5):1271-8. doi: 10.1097/01.asn.0000013883.99976.22.
- Kalliakmani P, Benou E, Goumenos DS. Cyclosporine A in adult patients with Henoch-Schönlein purpura nephritis and nephrotic syndrome; 5 case reports. Nephrol. 2011 Apr;75(4):380-3. doi: 10.5414/cn106553.
- Jauhola O, Ronkainen J, Autio-Harmainen H, Koskimies O, Ala-Houhala M, Arikoski P, et al. Cyclosporine A vs. methylprednisolone for Henoch-Schönlein nephritis: a randomized trial. Pediatric Nephrol. 2011 Dec;26(12):2159-66. doi: 10.1007/s00467-011-1919-5.
- Rostoker G, Desvaux-Belghiti D, Pilatte Y, Petit-Phar M, Philippon C, Deforges L, et al. High-dose immunoglobulin therapy for severe IgA nephropathy and Henoch-Schönlein purpura. Ann Intern Med 1994 Mar 15;120(6):476-84. doi: 10.7326/0003-4819-120-6-199403150-00005.
- Augusto JF, Sayegh J, Delapierre L, Croue A, Tollis F, Cousin M, et al. Addition of plasmapheresis to glucocorticosteroids for the treatment of severe Henoch-Schönlein purpura in adults: a case series. Am J Renal Disease. 2012 May;59(5):663-9. doi: 10.1053/j.ajkd.2011.12.015
- Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Working Group. 2021 KDIGO clinical practice guideline for the management of glomerular diseases. Kidney Int. 2021 Oct;100(4S):S1-S276. doi: 10.1016/j.kint.2021.05.021.
- KDIGO 2024 CLINICAL PRACTICE GUIDELINE FOR THE MANAGEMENT OF IMMUNOGLOBULIN A NEPHROPATHY (IgAN) AND IMMUNOGLOBULIN A VASCULITIS (IgAV)
- José Hernández-Rodríguez, Cristina Carbonell, José-A Mirón-Canelo, Sandra Diez-Ruiz, Miguel Marcos, Antonio J. Chamorro,Rituximab treatment for IgA vasculitis: A systematic review,Autoimmunity Reviews, Volume 19, Issue 4, 2020,102490, ISSN 1568-9972,https://doi.org/10.1016/j.autrev.2020.102490.
- ERVIOLI, Luisa Fernanda. Vasculitis por IgA del adulto. Revisión de la literatura. Rev. Urug. Med. Int., Montevideo , v. 9, e201, dic. 2024 .